





Coup de cœur RECHERCHE
Projecteur sur le docking moléculaire
Le docking moléculaire est une méthode informatique utilisée pour prédire la structure tridimensionnelle des complexes moléculaires, par la simulation de l’interaction entre une molécule cible, souvent une protéine, et un ligand, comme un médicament potentiel. En identifiant la position la plus favorable du ligand dans le site actif de la protéine cible, c’est-à dire la plus stable, le docking moléculaire aide à prédire les interactions moléculaires et à améliorer leur efficacité. En somme, il révèle la disposition spatiale optimale des molécules lorsqu’elles se lient pour former un complexe, comme illustré Figure 1.
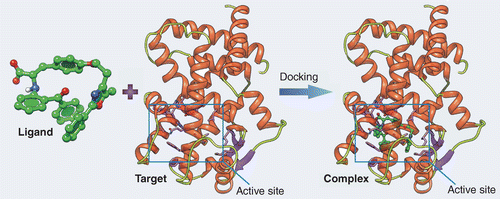
Figure 1 : principe du docking moléculaire, illustration de l’interaction entre une molécule (ligand) et une protéine (Target) au niveau de son site actif (Rectangle)
Depuis son développement dans les années 1990 pour la découverte de médicaments anti-VIH1, le docking moléculaire est devenu une méthode standard et essentielle en recherche pharmaceutique2 pour prédire les interactions protéine-ligand et aider la conception de médicaments. Ses applications s’étendent dans de nombreux autres domaines, quand l’optimisation de la disposition spatiale des macromolécules permet d’améliorer leur efficacité : en médecine personnalisée pour prédire la réponse des patients à certains médicaments en fonction de leur profil génétique et moléculaire, en industrie agroalimentaire pour optimiser des arômes et des additifs alimentaires, en agrochimie pour concevoir de nouveaux pesticides et herbicides plus spécifiques …
Quelques rappels sur la structure des protéines
Les protéines, molécules essentielles en tant que cibles thérapeutiques, possèdent des sites actifs qui permettent des interactions spécifiques avec d’autres molécules. Ces sites actifs correspondent à des régions particulières de la structure tridimensionnelle des protéines, où se déroulent des réactions chimiques ou des liaisons moléculaires. Leur structure précise permet une reconnaissance et une liaison sélective avec les molécules cibles, favorisant ainsi les fonctions biologiques. Les interactions au niveau du site actif sont clés, stabilisées par diverses forces telles que les liaisons hydrogènes et les interactions hydrophobes, comme illustré Figure 2.
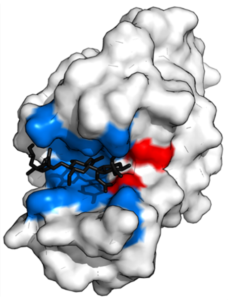
Figure 2 : représentation d’une protéine interagissant avec un ligand (molécule présentée en noir) au niveau de son site actif, illustré en rouge et en bleu4.
Pour comprendre le fonctionnement des protéines, il est important d’appréhender leur structure, organisée en quatre niveaux hiérarchiques intégrant les sites actifs comme éléments essentiels. La structure primaire, ou séquence, correspond à la succession linéaire des acides aminés sans référence à une configuration spatiale. La structure secondaire se focalise sur les motifs locaux de la chaîne polypeptidique, tels que l’α-hélice et les feuillets β, stabilisés par des liaisons hydrogènes entre les atomes d’oxygène et d’hydrogène. Une représentation visuelle de ces structures est présentée Figure 3.
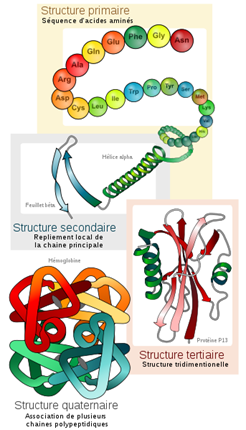
Figure 3 : représentation schématique des quatre niveaux hiérarchiques de la structure des protéines5.
Ces éléments structuraux secondaires s’associent pour former la structure tridimensionnelle de la protéine, connue sous le nom de structure tertiaire, qui est essentielle à sa fonction biologique et peut comporter des motifs et des domaines fonctionnels distincts. De plus, les protéines peuvent se combiner en complexes multimériques, définissant leur structure quaternaire, qui implique l’interaction de multiples sous-unités protéiques et joue un rôle crucial dans de nombreux processus cellulaires. La compréhension de ces concepts fondamentaux de la structure protéique, y compris les sites actifs et leur rôle dans les interactions moléculaires, est essentielle pour le développement du docking moléculaire. En effet, la méthode met en œuvre deux approches, basées respectivement sur l’étude de la complémentarité géométrique et chimique des surfaces, et le calcul de l’énergie du complexe macromoléculaire.
Différents types de docking selon le type d’interactions moléculaires
Le docking protéine-protéine, représenté Figure 4, se focalise sur les interactions entre deux protéines distinctes. Il est essentiel pour comprendre la formation de complexes protéiques impliqués dans divers processus cellulaires, tels que la régulation de la transcription et de la traduction6,7. Par exemple, dans la recherche sur le cancer, ce type de docking est utilisé pour étudier les interactions entre les protéines impliquées dans la régulation de la croissance cellulaire ou la progression tumorale, offrant ainsi des perspectives pour le développement de nouvelles thérapies ciblées8.
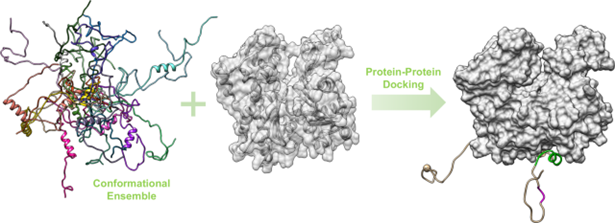
Figure 4 : représentation du docking entre deux protéines9.
Le docking protéine-peptide, représenté Figure 5, implique l’interaction entre une protéine et un peptide. Il est souvent utilisé pour étudier les mécanismes de régulation de la signalisation cellulaire10,11. Par exemple, il peut être appliqué à l’étude des interactions entre les protéines kinases et leurs peptides substrats, offrant des informations essentielles pour la conception de médicaments ciblant spécifiquement les voies de signalisation impliquées dans diverses maladies12.
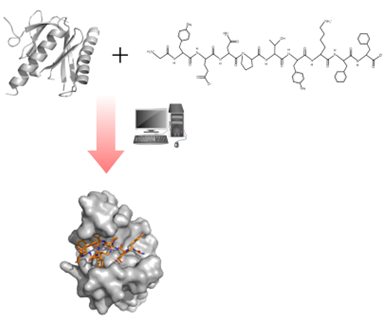
Figure 5 : illustration du docking entre une protéine (molécule à gauche) et un peptide (molécule à droite)13.
Enfin, le docking protéine-ligand concerne l’interaction entre une protéine et une petite molécule organique appelée ligand, également clé dans la découverte de médicaments3,14. Il permet de prédire comment les petites molécules interagissent avec leurs cibles protéiques, offrant des informations capitales pour concevoir des médicaments agissant de manière très spécifique, en réduisant les effets secondaires indésirables15.
Docking rigide ou flexible ?
Le choix entre les approches rigide ou flexible dépend de la nature du système biomoléculaire étudié et des objectifs de recherche spécifiques16. Le docking rigide est préféré lorsque les protéines et les ligands conservent leurs conformations initiales lors de la formation du complexe, pour les interactions stables avec peu de changements conformationnels17. Quand les interactions impliquent des changements conformationnels significatifs, le docking flexible permet de prendre en compte les mouvements conformationnels pour optimiser les interactions intermoléculaires et améliorer la précision des prédictions de liaison18.
La figure 6 représente schématiquement ces méthodes, dans le cas d’un docking entre une protéine et un ligand.
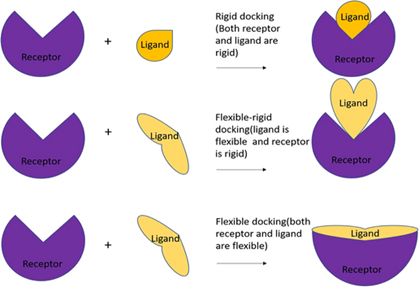
Figure 6 : approches rigide et flexible19
Quels logiciels ?
Les outils informatiques sont mobilisés pour prédire et modéliser les interactions entre différentes entités moléculaires, notamment les interactions des complexes protéine-ligand, afin de faciliter le processus de conception de médicaments en permettant aux chercheurs de prédire les structures tridimensionnelles des complexes. Parmi les logiciels, on peut citer, Haddock 20, Cluspro21 , LightDock 22,InterEvDock323, LZerD24 , AutoDock Vina25, et CABSDOCK26 .
L’intelligence artificielle (IA) apporte sa pierre à l’édifice en améliorant les performances du docking. Les méthodes basées sur l’apprentissage automatique, comme les réseaux de neurones, sont désormais largement utilisées. Par exemple, elles permettent de prédire avec précision les conformations des ligands et des protéines, ainsi que de filtrer les candidats de liaison potentiels. Ces avancées renforcent l’efficacité et la précision des simulations de Docking, ouvrant de nouvelles perspectives dans la découverte de médicaments. Parmi les logiciels qui intègrent ces techniques, citons AutoDock27, SWISS-MODEL28 et DeepDock29, qui offrent des solutions innovantes pour l’analyse moléculaire et la conception de médicaments.
Le docking moléculaire à l’EBI
Au sein de l’unité de recherche EBInnov®, deux projets exploitent ces techniques pour approfondir notre compréhension des processus biologiques et pour ouvrir de nouvelles perspectives thérapeutiques.
Tout d’abord, EBInnov® a développé un motif biomimétique inspiré de la fibronectine humaine, une glycoprotéine présente dans la matrice extracellulaire jouant un rôle clé dans l’adhésion cellulaire. Grâce à des techniques avancées de bio-informatique, notamment l’algorithme Alphafold30, la structure tridimensionnelle de ce motif a été déterminée. Cette résolution structurale a ensuite été utilisée pour explorer les interactions entre le motif et les protéines de la matrice extracellulaire, permettant une compréhension détaillée de leurs affinités et des différentes conformations possibles.
Ensuite, deuxième sujet faisant appel au docking, EBInnov® cherche à identifier de nouvelles cibles thérapeutiques dans la lutte contre les mélanomes. Ces cancers posent un défi majeur en raison de la résistance potentielle aux traitements immuno-thérapeutiques. Le projet vise à combiner des approches bio-informatiques, expérimentales et de modélisation moléculaire pour comprendre les mécanismes de résistance et identifier de nouvelles cibles thérapeutiques. Le docking moléculaire permet d’explorer les interactions entre les biomolécules impliquées dans le mélanome, ouvrant ainsi la voie à de nouvelles stratégies thérapeutiques.
Et demain ?
L’intégration croissante de l’intelligence artificielle va permettre d’améliorer encore l’efficacité des simulations, la compréhension des processus biologiques et d’accélérer le développement de nouvelles thérapies.
Contacts :
Dr Jad EID, Professeur de bioinformatique et biophysique à l’EBI
Références :
(1) Agu, P. C.; Afiukwa, C. A.; Orji, O. U.; Ezeh, E. M.; Ofoke, I. H.; Ogbu, C. O.; Ugwuja, E. I.; Aja, P. M. Molecular Docking as a Tool for the Discovery of Molecular Targets of Nutraceuticals in Diseases Management. Sci Rep 2023, 13 (1), 13398. https://doi.org/10.1038/s41598-023-40160-2.
(2) Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr Comput Aided Drug Des 2011, 7 (2), 146–157.
(3) Torres, P. H. M.; Sodero, A. C. R.; Jofily, P.; Silva-Jr, F. P. Key Topics in Molecular Docking for Drug Design. Int J Mol Sci 2019, 20 (18), 4574. https://doi.org/10.3390/ijms20184574.
(4) Liao, C.; Peach, M. L.; Yao, R.; Nicklaus, M. C. Molecular Docking and Structure-Based Virtual Screening. In In Silico Drug Discovery and Design; Future Science Book Series; Future Science Ltd, 2013; pp 6–20. https://doi.org/10.4155/ebo.13.181.
(5) Simon, M. Structures des protéines. Cours Pharmacie. https://www.cours-pharmacie.com/biochimie/structures-des-proteines.html (accessed 2024-02-08).
(6) Grosdidier, A. Conception d’un logiciel de docking et applications dans la recherche de nouvelles molécules actives.
(7) Kurcinski, M.; Kmiecik, S.; Zalewski, M.; Kolinski, A. Protein–Protein Docking with Large-Scale Backbone Flexibility Using Coarse-Grained Monte-Carlo Simulations. Int J Mol Sci 2021, 22 (14), 7341. https://doi.org/10.3390/ijms22147341.
(8) Férey, N.; Bouyer, G.; Martin, C.; Drif, A.; Bourdot, P.; Ammi, M.; Nelson, J.; Burkhardt, J.-M.; Autin, L. Docking de Protéines En Réalité Virtuelle. Une Approche Hybride et Multimodale. Revue des Sciences et Technologies de l’Information – Série TSI : Technique et Science Informatiques 2009, 28 (8), 983–1015. https://doi.org/10.3166/tsi.28.983-1015.
(9) BioCIS – Biomolécules : Conception, Isolement et Synthèse – Interactions Protéine-Ligand. https://www.biocis.universite-paris-saclay.fr/?Interaction-Prot-Prot-900 (accessed 2024-02-08).
(10) Kurcinski, M.; Badaczewska‐Dawid, A.; Kolinski, M.; Kolinski, A.; Kmiecik, S. Flexible Docking of Peptides to Proteins Using CABS‐dock. Protein Sci 2020, 29 (1), 211–222. https://doi.org/10.1002/pro.3771.
(11) Chevrollier, N. Développement et application d’une approche de docking par fragments pour modéliser les interactions entre protéines et ARN simple-brin.
(12) Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Peptides Targeting Protein Kinases: Strategies and Implications. Physiology 2006, 21 (6), 411–418. https://doi.org/10.1152/physiol.00022.2006.
(13) Protein–Peptide Docking – Profacgen. https://www.profacgen.com/protein-peptide-docking.htm (accessed 2024-02-08).
(14) Hernández-Santoyo, A.; Tenorio-Barajas, A. Y.; Altuzar, V.; Vivanco-Cid, H.; Mendoza-Barrera, C.; Hernández-Santoyo, A.; Tenorio-Barajas, A. Y.; Altuzar, V.; Vivanco-Cid, H.; Mendoza-Barrera, C. Protein-Protein and Protein-Ligand Docking. In Protein Engineering – Technology and Application; IntechOpen, 2013. https://doi.org/10.5772/56376.
(15) Grosdidier, A. Conception d’un logiciel de docking et applications dans la recherche de nouvelles molécules actives.
(16) Agrawal, P.; Singh, H.; Srivastava, H. K.; Singh, S.; Kishore, G.; Raghava, G. P. S. Benchmarking of Different Molecular Docking Methods for Protein-Peptide Docking. BMC Bioinformatics 2019, 19 (13), 426. https://doi.org/10.1186/s12859-018-2449-y.
(17) Ketata, M. A.; Laue, C.; Mammadov, R.; Stark, H.; Wu, M.; Corso, G.; Marquet, C.; Barzilay, R.; Jaakkola, T. S. DiffDock-PP: Rigid Protein-Protein Docking with Diffusion Models; 2023.
(18) de Oliveira, E. B. Simulations moléculaires appliquées à l’acétylation de flavonoïdes catalysée par des lipases: influence des structures de la lipase et du flavonoïde et sur la régiosélectivité de la bioconversion.
(19) Mohanty, M.; Mohanty, P. S. Molecular Docking in Organic, Inorganic, and Hybrid Systems: A Tutorial Review. Monatsh Chem 2023, 154 (7), 683–707. https://doi.org/10.1007/s00706-023-03076-1.
(20) van Zundert, G. C. P.; Rodrigues, J. P. G. L. M.; Trellet, M.; Schmitz, C.; Kastritis, P. L.; Karaca, E.; Melquiond, A. S. J.; van Dijk, M.; de Vries, S. J.; Bonvin, A. M. J. J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. Journal of Molecular Biology 2016, 428 (4), 720–725. https://doi.org/10.1016/j.jmb.2015.09.014.
(21) Desta, I. T.; Porter, K. A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28 (9), 1071-1081.e3. https://doi.org/10.1016/j.str.2020.06.006.
(22) Jiménez-García, B.; Roel-Touris, J.; Barradas-Bautista, D. The LightDock Server: Artificial Intelligence-Powered Modeling of Macromolecular Interactions. Nucleic Acids Res 2023, 51 (W1), W298–W304. https://doi.org/10.1093/nar/gkad327.
(23) Quignot, C.; Postic, G.; Bret, H.; Rey, J.; Granger, P.; Murail, S.; Chacon, P.; Andreani, J.; Tuffery, P.; Guerois, R. InterEvDock3: A Combined Template-Based and Free Docking Server with Increased Performance through Explicit Modeling of Complex Homologs and Integration of Covariation-Based Contact Maps. Nucleic Acids Research 2021, 49. https://doi.org/10.1093/nar/gkab358.
(24) Christoffer, C.; Bharadwaj, V.; Luu, R.; Kihara, D. LZerD Protein-Protein Docking Webserver Enhanced With de Novo Structure Prediction. Frontiers in Molecular Biosciences 2021, 8.
(25) Martz, F. Développement d’une nouvelle méthode de docking basée sur les mécanismes enzymatiques et guidée par des groupes prosthétiques.
(26) Kurcinski, M.; Pawel Ciemny, M.; Oleniecki, T.; Kuriata, A.; Badaczewska-Dawid, A. E.; Kolinski, A.; Kmiecik, S. CABS-Dock Standalone: A Toolbox for Flexible Protein-Peptide Docking. Bioinformatics 2019, 35 (20), 4170–4172. https://doi.org/10.1093/bioinformatics/btz185.
(27) Grosdidier, A. Conception d’un logiciel de docking et applications dans la recherche de nouvelles molécules actives.
(28) Mezhoud, K. Docking Principes et méthodes.
(29) Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M. E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6 (6), 939–949. https://doi.org/10.1021/acscentsci.0c00229.
(30) Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2.

